熱線:021-66110810,66110819
手機:13564362870
熱線:021-66110810,66110819
手機:13564362870
經2-DG處理的大鼠腦切片的病理變化
圖3經2-DG處理的大鼠海馬體外切片和組織樣本的代謝和電參數變化。A:對長期接受2-DG處理的動物海馬切片進行的體內外測量顯示,與接受載體處理的動物切片相比,它們的神經元對葡萄糖剝奪的耐受性要差得多。B:在經2-DG處理的大鼠切片中(齒狀回的通道記錄),Em沒有變化(n=29,P>0.2),而EGABA則顯著去極化(P<0.01,n=20)。
對長期接受2-DG處理的動物海馬切片進行的體內外測量顯示,與接受藥物處理的動物切片相比,它們的神經元對葡萄糖剝奪(低葡萄糖溶液,0.1mM)的耐受性要差得多(圖3A)。2-DG組大鼠的eLFP積分衰減到50%的時間是經空白載體處理的大鼠的一半(約25分鐘,n==11,與>70分鐘,n=12;P<0.001),這可能是因為糖原儲存減少的緣故(圖3C)。以前也曾使用類似的方案來確定糖原儲備及其對神經元活動的支持。
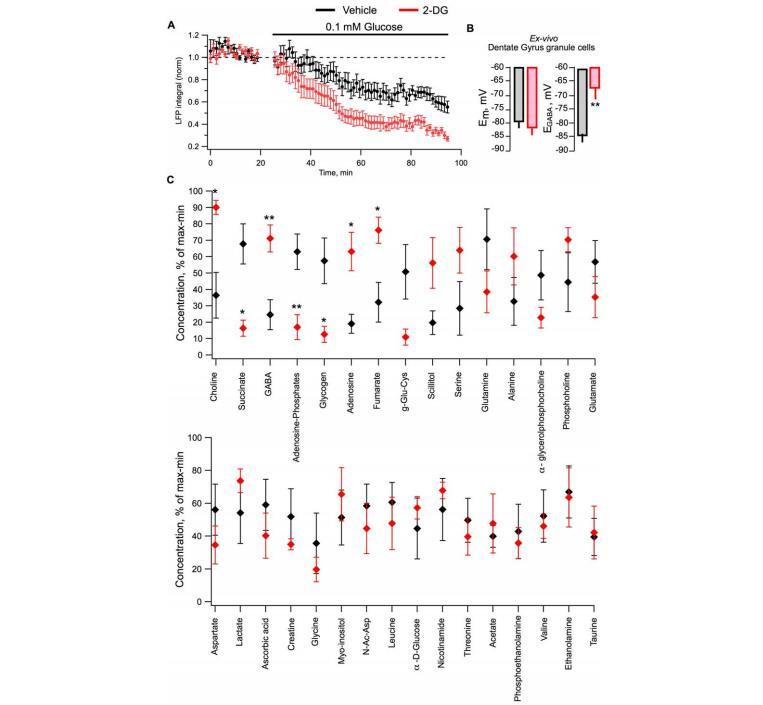
圖3 C:腦組織樣本的核磁共振分析結果(對照組[載體]大鼠,n=5;2-DG大鼠,n=5)。N-AcAsp表示N-乙酰天冬氨酸。
在體外切片中對單個神經元(齒狀回顆粒細胞)的測量也表明,雖然Em沒有變化(-79.3±2.3和-82±2.4mV,n=29,P>0.2),但在經2-DG處理的大鼠神經元中,EGABA明顯去極化(-67.2±4.0 vs.-83.7±3.4mV,n=29,P<0.01;圖3B),這表明抑制性驅動力的效力大大降低。這可能是2-DG治療期間網絡過度興奮性發展的一種細胞機制。
對2-DG處理過的大鼠腦樣本進行核磁共振分析后發現,它們的腦能量儲備(糖原和腺苷酸減少,腺苷增加)和線粒體代謝(琥珀酸減少)發生了顯著的有害變化(圖3C)。
討論
我們的研究結果表明,慢性代謝低下是后天性癲癇的公認風險因素(如腦外傷、中風、病毒感染、癲癇狀態)的特征,特別是葡萄糖利用率降低(通常在癲癇患者中觀察到),很容易引發癲癇。反過來,我們最近發現,癲癇發作本身會導致葡萄糖利用率快速而持久地下降,從而完成癲癇誘發和發展的惡性循環。我們提出,與氧化應激和神經炎癥相關的能量供應不足是后天癲癇發生的誘因和驅動力。本研究的結果提供了與風險因素相關的腦能量代謝不足與癲癇發生之間缺失的聯系。
關于癲癇治療,我們的研究結果表明,長期抑制大腦能量代謝是一種危險的策略,因為這會進一步加劇癲癇發生過程中已經形成的代謝危機。這種對能量供應的抑制不太可能像有些人認為的那樣,有助于觀察到KD的抗癲癇作用,因為據報道,在KD期間ATP合成正常或增加,而腦組織中的ATP水平要么保持不變,要么增加。據報道,無論酮病或非酮病狀態如何,KD期間葡萄糖的氧化能力都很高,盡管也發現腦葡萄糖代謝率降低。我們的結論是,在KD期間,由于酮體部分替代葡萄糖為線粒體提供燃料,糖酵解可能會有所減少;但這不應被視為能量代謝的全面抑制。
以前的研究曾試圖通過在點燃程序之前立即靜脈注射2-DG來抑制糖酵解,從而復制KD的抗癲癇效應。作者們的研究表明,在不同的點燃方法和2-DG劑量(75-500毫克/千克)下,點燃效率和由此導致的癲癇都會明顯降低。然而,如果對血液和大腦中的2-DG濃度進行估算,就很難支持這一結論。一項在大鼠身上進行的研究顯示,2-DG可預防缺血引起的死亡,該研究使用了1.6克/千克的靜脈注射量,結果顯示血液中的2-DG含量高達1.6毫摩爾。假設2-DG和葡萄糖通過血腦屏障的轉運相似,且葡萄糖梯度為5:1(血液中6mM對大腦ECF中1.3mM),則大腦中的2-DG濃度為0.32mM。這種計算方法可能在一定程度上低估了2-DG的水平,因為低ECF葡萄糖可能是通過有效的細胞消耗來維持的。然而,耐人尋味的是,注射2-DG還會導致血糖水平和腦血流量同時顯著上升,這在動物和人類身上都有報道。這些因素應導致大腦ECF葡萄糖的相應增加。使用與我們的"方法"中相同的糖酵解抑制計算方法,并考慮到由此得出的腦2-DG和葡萄糖濃度,我們可以估計,即使使用用于抗癲癇治療的最高2-DG劑量(500毫克/千克),腦糖酵解抑制作用也只能達到約1%至2%,幾乎可以忽略不計。因此,皮下注射2-DG更有可能是通過全身驅動效應來發揮抗癲癇作用的,例如上述腦葡萄糖供應量和腦血流量的短暫增加。在我們的活體實驗中,腦室內注射2-DG所使用的濃度應能短暫抑制腦糖酵解約14%。這樣的量級不太可能造成大規模的形態學破壞或細胞死亡,因此我們得出結論,是對腦糖酵解的直接和部分抑制導致了所觀察到的癲癇表型。事實上,單次腦室內注射2-DG已被用于研究葡萄糖剝奪對大鼠攝食行為的影響,其濃度比我們的濃度高出10倍以上,這表明腦室內給藥在腦細胞存活和糖酵解抑制方面是有效的。否則,鑒于2-DG治療的慢性性質以及腦內給藥方法,很難將我們的活體研究結果與之前的研究結果進行直接比較。
最近一項使用LDH阻斷劑作為能量代謝抑制替代方法的研究報告稱,該方法具有顯著的急性抗驚厥效果。研究表明,通過腹腔注射或海馬內注射LDH阻斷劑抑制糖酵解,可迅速降低注射凱因酸鹽的小鼠海馬中的高壓尖峰頻率。作為LDH阻斷劑作用的一種可能機制,作者提出LDH抑制會導致星形膠質細胞乳酸生成和釋放受阻,從而造成神經元能量短缺,激活通過KATP通道的超極化電流,消除網絡的過度興奮性。
然而,該研究中的所有體外記錄都是在室溫和含有2.5毫摩爾葡萄糖的ACSF中進行的,作者認為這是一個"類似于體內"的濃度。同時,體外切片和體內的葡萄糖消耗量也大不相同。在切片中,由于靜息狀態下的大量消耗,組織內的基礎葡萄糖濃度幾乎是灌流液的一半(見圖4A)。在突觸激活過程中,細胞外葡萄糖和氧氣水平會下降,并在刺激后緩慢恢復。相反,在活體大腦中,網絡激活過程中消耗的葡萄糖和氧氣會迅速從血管中補充回來(見圖4B)。因此,在ACSF中含有2.5毫摩爾葡萄糖的情況下,切片組織中的葡萄糖濃度即使在靜止時也只有約1毫摩爾,這當然是一種缺糖狀態,而在突觸刺激過程中葡萄糖濃度會進一步降低。
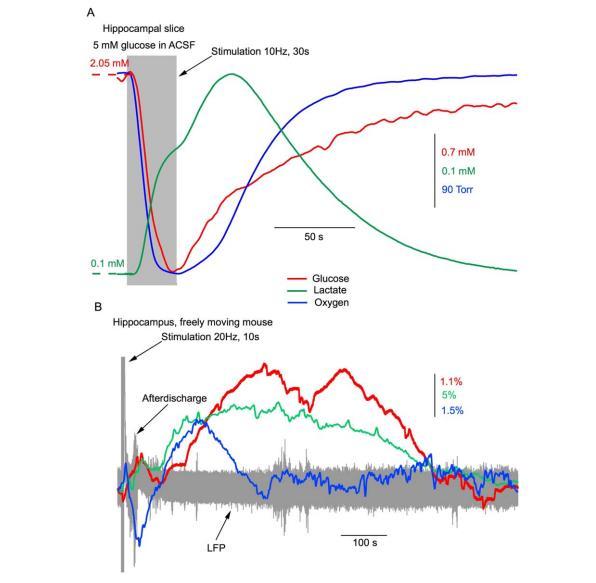
對體內結果的解釋源于一個假設,即乳酸從星形膠質細胞單向流入神經元并被用作能量燃料。然而,我們在切片和體內對乳酸釋放的記錄并沒有顯示網絡激活引起大量乳酸消耗的跡象--恰恰相反,我們記錄到的是大量乳酸釋放(見圖4)。盡管乳酸肯定可以用作能量來源,但在正常情況下,神經元中的部分丙酮酸似乎會被LDH轉化為乳酸并釋放到細胞外空間。如果是這樣的話,那么阻斷LDH確實可能會通過丙酮酸誘導的糖酵解抑制而導致嚴重的能量剝奪。例如,Sada等人的研究表明,抑制LDH會導致興奮性突觸后電流減少70%,這很可能是由于突觸遞質釋放減少所致(因為突觸后靶細胞是在細胞內溶液中含有4mMATP的全細胞構型下記錄的,這就避免了LDH受阻導致的能量短缺)。突觸前機制可能主要依賴于糖酵解提供的ATP,因此報告的LDH效應很可能是突觸前糖酵解抑制的結果。因此,與2-DG的急性效應類似,能量剝奪引起的最初網絡活動減少也會導致短期抗癲癇效應,盡管不能排除相反的癲癇發作效應。我們的結果還表明(見圖1A),能量缺乏可能會導致雙向效應,這取決于誘發的突觸傳遞下調與Em和EGABA去極化之間的平衡。
總之,這項研究的結果支持了我們關于后天性癲癇是一種代謝性疾病的概念觀點,突出了抑制腦能量代謝作為一種治療策略的風險。
 相關新聞
相關新聞