熱線:021-66110810,66110819
手機:13564362870
熱線:021-66110810,66110819
手機:13564362870
研究簡介:大腦雖然只占體重2%,卻消耗了近20%的靜息能量,其ATP幾乎全部由葡萄糖有氧氧化供給。幾十年來,神經代謝領域卻流傳著一個看似矛盾的假設:當神經元突然高頻放電時,第一時間并不動用“主發電機”——線粒體氧化磷酸化,而是先啟動“應急電池”——糖酵解,尤其是在星形膠質細胞內的糖酵解。本研究利用大鼠海馬腦片,結合氧微電極記錄、NADH熒光成像及數學建模,系統評估了神經元活動瞬間的能量來源。傳統觀點認為,神經活動首先由糖酵解(主要在星形膠質細胞)供能,再通過乳酸轉運給神經元進行氧化磷酸化。研究卻發現在阻斷乳酸脫氫酶(LDH)、或僅用20%O?模擬生理低氧條件下,神經元刺激仍迅速降低胞外O?和胞內NADH,提示氧化磷酸化被立即動員.藥理學逐步阻斷突觸前動作電位、Ca2?內流-遞質釋放、突觸后谷氨酸受體電流及動作電位,可分別減少11%、17%、46%、26%的O?消耗,證明這些關鍵信息處理環節均由氧化磷酸化直接供能.阻斷LDH后,活動誘發的O?與NADH變化幅度不變,否定了星形膠質細胞-神經元乳酸穿梭假說對瞬時能量供給的必要性。該工作顛覆了“先糖酵解后氧化”的傳統教條,明確了大腦信息處理主要由氧化磷酸化實時驅動,為理解腦能量代謝及fMRI信號生理基礎提供直接證據。
Unisense微電極系統的應用
使用了Unisense Clark型玻璃氧微電極,尖端直徑10μm。實現了原位測量海馬切片內胞外氧濃度變化,作為氧化磷酸化速率的直接指標。微電極尖端定位于CA1輻射層距錐體細胞層30μm、距切片表面約100μm處。通過Clark型玻璃微電極實現10μm空間分辨率,實時監測刺激誘發的O?濃度動態變化。量化不同藥物(如NBQX、Cd2?)對O?消耗的抑制比例(如突觸后機制占72%),為能量分配提供實驗依據。
實驗結論
研究通過海馬腦片多手段實驗,首次直接證明神經元活動瞬間所需ATP主要由氧化磷酸化即時提供,而非傳統認為的“先糖酵解后氧化”或星形膠質細胞-神經元乳酸穿梭模式。阻斷乳酸脫氫酶或降低氧濃度后,神經元刺激仍迅速耗氧并降低NADH,表明氧化磷酸化被立即動員,承擔≥62%的ATP增量。利用藥理-建模拆分,11%的氧耗用于突觸前動作電位,17%用于Ca2?內流-遞質釋放,46%用于突觸后谷氨酸受體電流,26%用于突觸后動作電位。抑制LDH不改變刺激誘發的氧耗與NADH變化,否定了星形膠質細胞產乳酸快速供神經元氧化磷酸化的必要性。
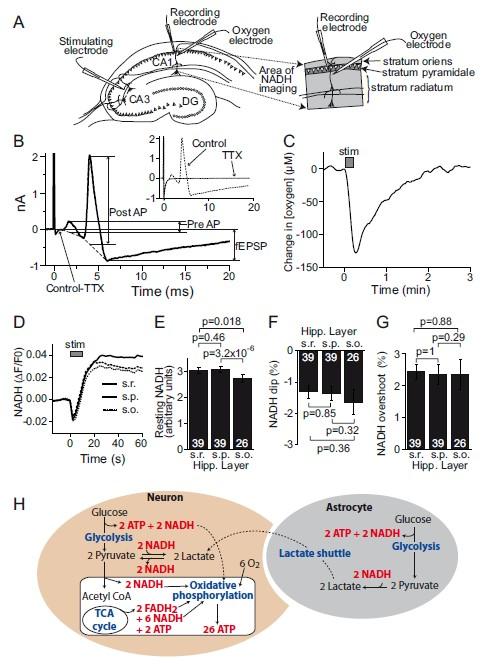
圖1、刺激誘發的場電位及O?與NADH變化。A圖:實驗電極位置及成像區域示意圖。B圖:左上小圖:0.33 Hz刺激下記錄的場電位(含突觸前動作電位、場興奮性突觸后電位fEPSP及突觸后群體峰電位),TTX阻斷后僅保留刺激偽跡。主圖:TTX阻斷前后的差值曲線,顯示各成分測量方法(突觸前動作電位幅度、fEPSP斜率、群體峰電位幅度)。C圖:20 Hz刺激10秒誘發細胞外[O?]下降(均值118±8μM)。D圖:NADH熒光雙相響應(初始下降1.4%,后續超調2.4%),在輻射層(s.r.)、錐體層(s.p.)和腔隙層(s.o.)無顯著差異。E-G圖:各層NADH靜息熒光(E)、刺激誘發的NADH下降(F)及超調(G)的定量比較。H圖:糖酵解、TCA循環與氧化磷酸化對NADH水平的調控機制示意圖。
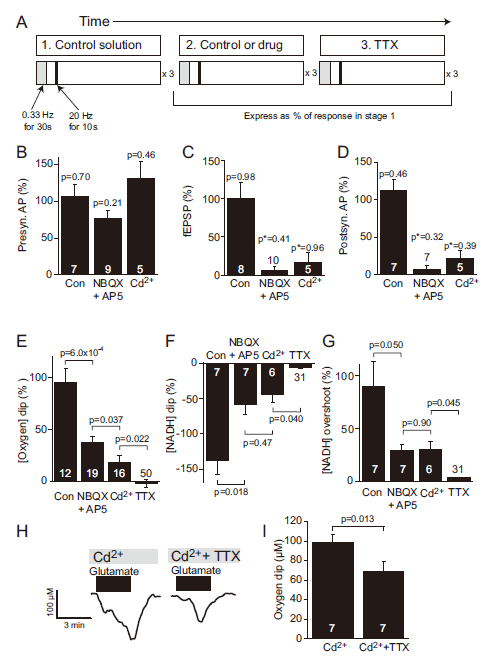
圖2、不同亞細胞機制的O?消耗藥理學分析。A圖:實驗流程設計(三階段刺激協議,含藥物干預)。B-D圖:NBQX+AP5(阻斷突觸后電流)選擇性抑制fEPSP和群體峰電位,對突觸前動作電位無影響。Cd2?阻斷突觸傳遞但保留突觸前動作電位,TTX阻斷全部電活動。E-G圖:逐步阻斷突觸后活動(NBQX+AP5)、遞質釋放(Cd2?)及全部活動(TTX)使[O?]下降依次減少63%、82%和100%。NADH下降及超調呈現類似階梯式抑制。H-I圖:谷氨酸(500μM)在Cd2?存在下誘發的[O?]下降,TTX進一步減少29%,表明動作電位消耗O?
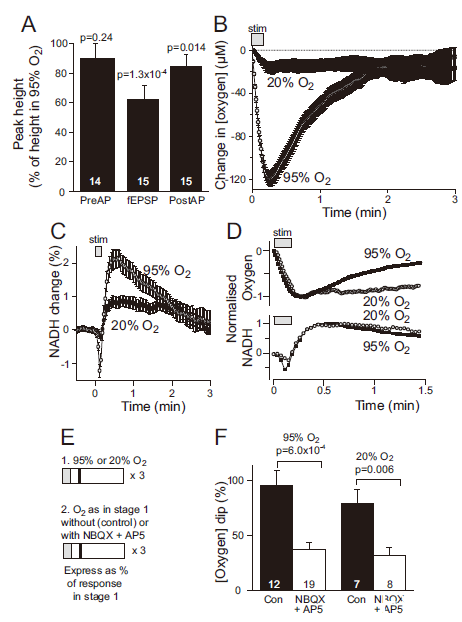
圖3、生理O?水平下氧化磷酸化支持神經活動。A圖:20%O?灌流降低fEPSP(38%)和群體峰電位5%),但不影響突觸前動作電位。B圖:20%O?下[O?]下降幅度減小但持續時間延長(恢復需5分鐘)。C圖:NADH初始下降及超調幅度在低O?下降低。D圖:標準化后的O?下降與NADH超調時間重疊。E-F圖:NBQX+AP5在95%和20%O?下對[O?]下降的抑制比例無差異(63%vs.68%),表明突觸后O?消耗與O?水平無關。
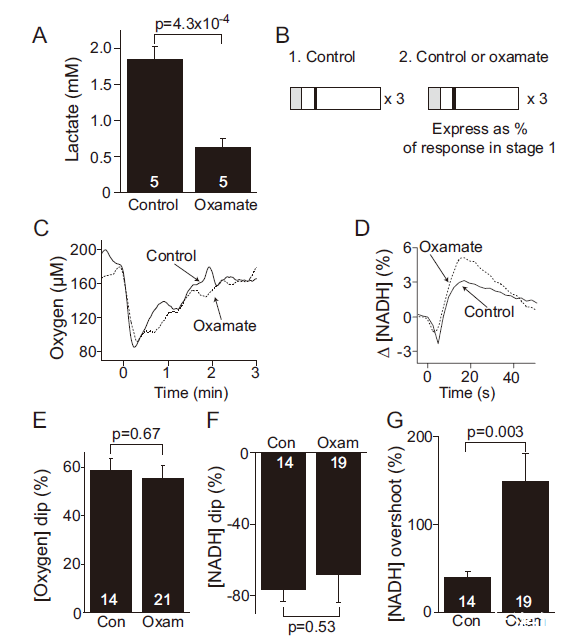
圖4、LDH阻斷不影響O?與NADH初始下降。A圖:LDH抑制劑草酸鹽使細胞外乳酸減少66%。B圖:實驗設計(階段2加入oxamate)。C-D圖:oxamate不改變[O?]下降(E)和NADH初始下降(F),但增加NADH超調(G)(p=0.003),提示糖酵解延遲激活。
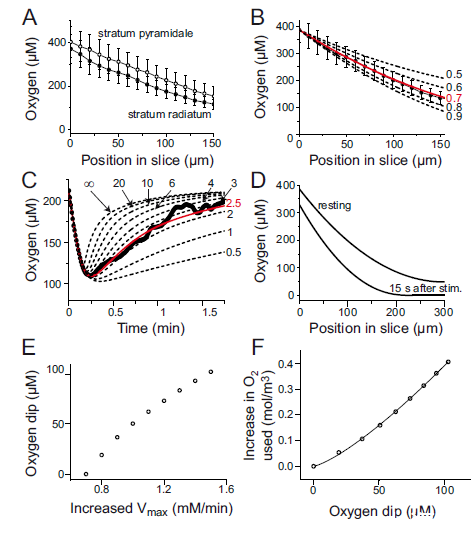
圖5、巨噬細胞復極化的機制研究。a)接受不同處理的RAW 264.7細胞的RNA測序分析示意圖。該圖由Figdraw創建。b)來自不同群體的RAW 264.7細胞基因的PCA分析。數據按原樣顯示(N=3)。c)來自不同組的RAW 264.7細胞轉錄組中差異表達基因的基因聚集。數據按原樣顯示,并按平均值顯示(N=3)。d)來自不同組的RAW 264.7細胞的炎癥相關途徑中涉及的DEGs的熱圖分布。數據按原樣顯示(N=3)。e)LPS/IFN-γ和LPS/IFN-γ+CSN處理的RAW 264.7細胞轉錄組中DEGs的火山圖。f)LPS/IFN-γ和LPS/IFN-γ+CSN處理細胞的前15個DEGs的相互作用。g)LPS/IFN-γ和LPS/IFN-γ+CSN處理細胞的KEGG富集分析氣泡圖h)Nfkb2的mRNA表達,來自不同組的RAW 264.7細胞的Mapk12和Tnf。數據表示為SD±均值(N=3)。i)IL-17信號通路的GSEA圖顯示,用LPS/IFN-γ處理的RAW 264.7細胞CSN處理后顯著下調(NES=-1.74)。j)CSN巨噬細胞復極化的分子機制示意圖。k)來自不同組的RAW 264.7細胞的p65、p38和FosB的蛋白質印跡分析。
結論與展望
本研究研究了大腦信息處理過程中能量供應的核心機制,挑戰了"神經元活動初期依賴糖酵解供能"的傳統觀點。通過海馬體切片實驗,研究人員驗證了氧化磷酸化(而非糖酵解)是突觸前動作電位、神經遞質釋放、突觸后電流和動作電位等關鍵神經活動的主要能量來源。本研究首次整合Unisense O?電極、NADH成像和場電位記錄,實現多參數代謝-電活動同步分析。通過Clark型玻璃微電極實現10μm空間分辨率,實時監測刺激誘發的O?濃度動態變化。這一發現對理解腦功能成像(如BOLD信號)的生理基礎具有重要意義。本顛覆了“糖酵解先供能”的傳統教條,為理解大腦信息處理的能量供應機制、解釋BOLD-fMRI信號生理基礎提供了直接實驗依據。
 相關新聞
相關新聞