熱線:021-66110810,66110819
手機:13564362870
熱線:021-66110810,66110819
手機:13564362870
Aβ initiates brain hypometabolism and network dysfunction via NOX2 activation: a potential onset mechanism of Alzheimer’s disease
Aβ 誘導急性氧化應激,導致阿爾茨海默病發病機制中的腦葡萄糖代謝減退和多動癥
來源:biorxiv 2020.08.12.248492v4;Communications Biology volume 4, Article number: 1054 (2021)
1. 論文摘要核心內容
研究揭示β-淀粉樣蛋白(Aβ????)通過激活NADPH氧化酶2(NOX2)誘發腦葡萄糖低代謝和神經網絡功能障礙的機制:
核心發現:
Aβ????激活NOX2 → 誘發氧化應激 → 抑制糖酵解 → 導致腦葡萄糖利用率下降(圖1)。
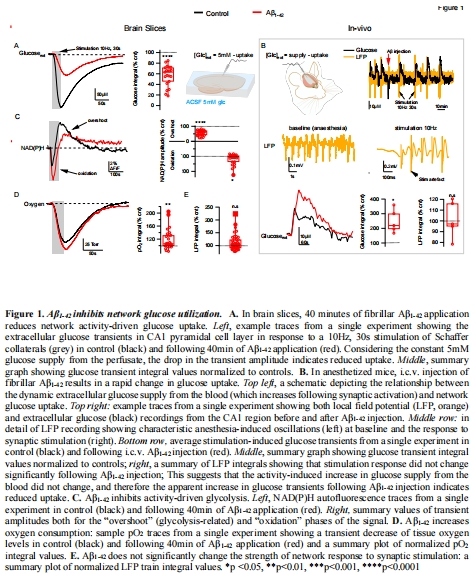
葡萄糖低代謝進一步引發海馬網絡超興奮(圖3)、病理性高頻振蕩(pHFOs)及焦慮/攻擊行為(圖4)。
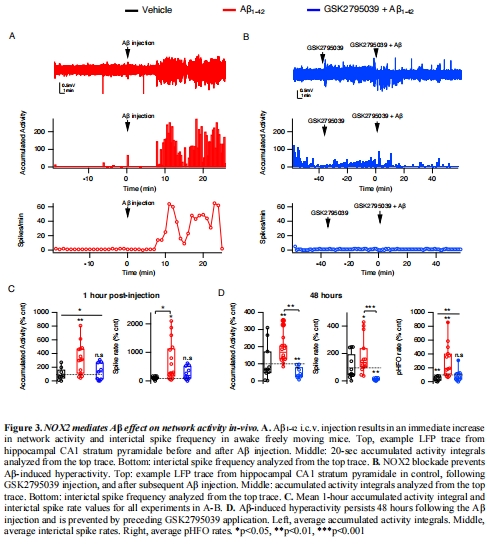
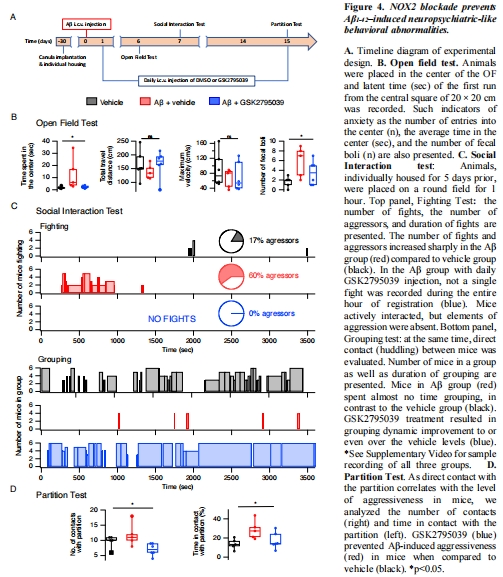
NOX2抑制劑GSK2795039可完全阻斷上述病理改變(圖2-4)。
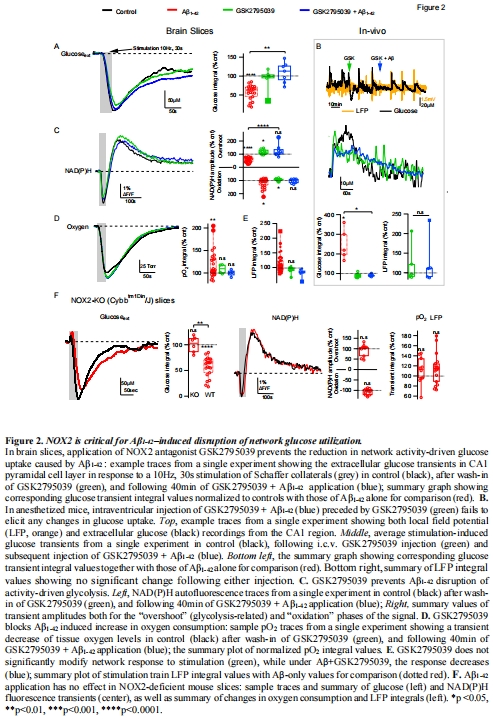
意義:首次明確NOX2是連接Aβ毒性、腦低代謝與神經功能障礙的關鍵節點,為阿爾茨海默病(AD)早期干預提供新靶點。
2. 研究目的
闡明AD早期葡萄糖低代謝的分子機制(Aβ是否通過NOX2誘發氧化應激)。
驗證NOX2激活如何導致神經網絡異常(超興奮、癲癇樣活動)。
探究NOX2抑制劑能否逆轉Aβ誘導的行為學異常(焦慮、攻擊性)。
3. 研究思路
1.模型建立:
離體實驗:小鼠海馬腦片施加Aβ????(400 nM),模擬AD病理環境(圖1)。
在體實驗:清醒/麻醉小鼠腦室內(i.c.v.)注射Aβ????(圖1B, 圖3)。
2.干預設計:
NOX2抑制劑GSK2795039預處理(圖2)。
NOX2基因敲除小鼠(Cybb<sup>tm1din/J</sup>)驗證(圖2F)。
3.多維度檢測:
代謝指標:葡萄糖/乳酸攝取、NAD(P)H/FAD熒光(糖酵解)、氧耗(圖1)。
電生理:局部場電位(LFP)、病理性高頻振蕩(pHFOs)(圖3)。
行為學:曠場實驗(焦慮)、社交互動測試(攻擊性)(圖4)。
4. 關鍵數據及研究意義
(1) Aβ誘發葡萄糖低代謝(圖1)
數據:
Aβ????使腦片葡萄糖攝取↓57.7%(圖1A)、在體葡萄糖瞬態振幅↑2倍(反映攝取↓)(圖1B)。
NAD(P)H"超射"(糖酵解標志)↓ + "氧化相"↑(圖1C),氧耗↑(圖1D)。
意義:Aβ通過抑制糖酵解(非葡萄糖轉運)引發能量危機,線粒體代償性耗氧增加。
(2) NOX2的核心作用(圖2)
數據:
GSK2795039或NOX2基因敲除完全阻斷Aβ的代謝抑制(圖2A-F)。
Aβ升高脂質過氧化標志MDA(表1),GSK2795039逆轉此效應。
意義:NOX2介導的氧化應激是Aβ毒性的直接原因。
(3) 網絡功能障礙(圖3)
數據:
Aβ注射后1小時:LFP累積活動↑60.8%(圖3C),癲癇樣放電率↑325%(表1)。
48小時后:pHFOs(250-600 Hz)↑304%(圖3D)。
GSK2795039預防所有異常(圖3B-D)。
意義:代謝障礙→神經網絡超興奮→癲癇樣活動,NOX2是核心驅動因子。
(4) 行為學異常(圖4)
數據:
曠場實驗:Aβ組中央停留時間↑2倍(焦慮)、排便量↑(圖4B)。
社交測試:攻擊行為↑(打斗次數/持續時間↑)、群體親和行為↓(圖4C)。
隔板測試:接觸隔板時間↑(攻擊傾向)(圖4D)。
GSK2795039完全逆轉所有行為異常(圖4B-D)。
意義:Aβ通過NOX2誘發AD早期神經精神癥狀(焦慮、攻擊性)。
5. 研究結論
1.NOX2是AD起始關鍵節點:Aβ???? → 激活NOX2 → 氧化應激 → 抑制糖酵解 → 腦葡萄糖低代謝。
2.代謝-神經功能耦聯:葡萄糖低代謝 → 神經網絡超興奮 → 癲癇樣放電及行為異常。
3.治療靶點驗證:NOX2抑制劑GSK2795039阻斷上述病理級聯,具臨床轉化潛力。
4.修正AD模型:早期AD以糖酵解障礙為主(線粒體功能尚存),顛覆傳統"線粒體衰竭"假說。
6. 丹麥Unisense電極的核心價值
(1) 技術優勢
多參數實時同步監測:
氧分壓(pO?)電極:直接量化組織氧耗(圖1D),發現Aβ誘導代償性耗氧↑。
葡萄糖/乳酸酶電極:實時捕捉刺激后代謝瞬態變化(圖1A-B),證實葡萄糖攝取↓早于電活動改變。
高時空分辨率:
電極尖端直徑25μm(方法部分),實現細胞微環境動態追蹤(如CA1區糖酵解瞬時抑制)。
(2) 顛覆性發現
1.揭示"代謝悖論":
Unisense數據首次證實:
Aβ抑制糖酵解(NAD(P)H超射↓)但增強氧化磷酸化(氧耗↑)(圖1C-D)。
→ 修正AD"線粒體功能障礙"傳統觀點,提出"糖酵解優先受損"新機制。
2.明確NOX2的核心地位:
通過抑制NOX2(GSK2795039)或基因敲除(Cybb<sup>tm1din/J</sup>),完全逆轉Aβ的代謝抑制效應(圖2)。
→ 為抗氧化治療提供精準靶點(非廣譜抗氧化劑)。
(3) 臨床轉化意義
早期診斷標志物:
刺激后葡萄糖瞬態振幅變化(圖1B)可作為AD超早期無創生物標志物。
治療評估工具:
Unisense實時監測GSK2795039的代謝保護效果(圖2),加速藥物臨床轉化。
機制驅動療法:
否定單純抗氧化策略(無效臨床試驗),確立NOX2特異性抑制為AD預防新方向。
核心圖示:Aβ-NOX2-低代謝級聯
Aβ???? → 激活NOX2 → 氧化應激 → 抑制糖酵解 → 葡萄糖低代謝
↓
能量危機 → 神經網絡超興奮 → 癲癇樣放電/行為異常
↓
NOX2抑制劑(GSK2795039)阻斷全程
總結:
本研究通過整合離體腦片-在體電生理-行為學多維模型,揭示NOX2是Aβ誘發腦低代謝的核心機制。丹麥Unisense電極的高精度代謝監測能力為發現"糖酵解特異性抑制"提供關鍵技術支撐,推動AD早期診斷與靶向NOX2的精準治療策略。