熱線:021-66110810,66110819
手機:13564362870
熱線:021-66110810,66110819
手機:13564362870
Thyroid hormone increases oxygen metabolism causing intrarenal tissue hypoxia; a pathway to kidney disease
甲狀腺激素增加氧代謝,導致腎內組織缺氧;導致腎臟疾病的途徑
來源: PLoS ONE 17(3): e0264524
一、摘要概述
本研究通過三碘甲狀腺原氨酸(T3)誘導腎臟氧耗增加,探究腎組織缺氧本身是否直接導致腎病:
核心發現:
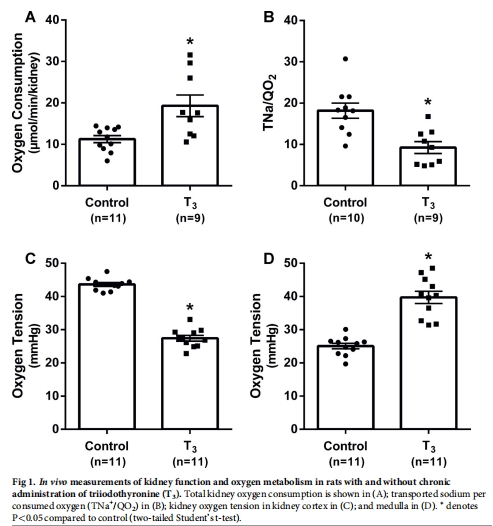
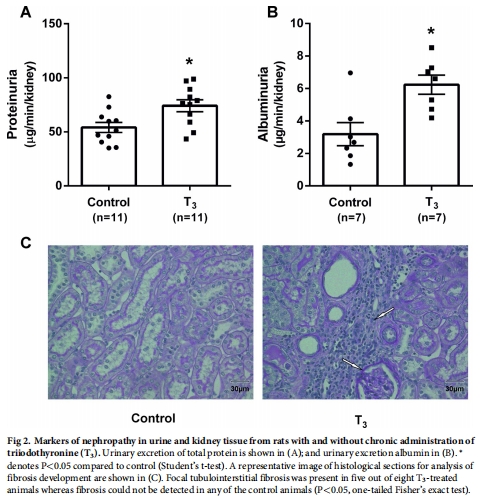
T3處理7周顯著增加大鼠腎臟總氧耗(QO?),導致腎皮質缺氧(圖1C),并誘發腎病(蛋白尿、腎小管間質纖維化)(圖2)。
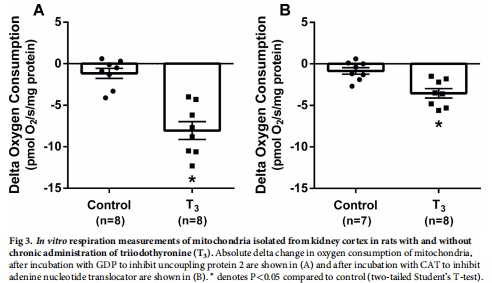
機制上,T3通過線粒體解偶聯(UCP-2和ANT介導)增加氧耗(圖3),且該過程獨立于高血壓、高血糖和氧化應激(表2)。
創新點:首次通過甲狀腺激素模型排除混雜因素(如糖尿病、高血壓),直接證明腎組織缺氧是腎病的獨立致病通路。
二、研究目的
解決兩大科學問題:
驗證缺氧假說:在排除高血糖、高血壓等混雜因素后,明確腎組織缺氧是否直接導致腎病。
揭示機制:探究T3增加腎氧耗的具體分子機制(尤其線粒體功能)。
三、研究思路
采用體內-體外結合的實驗設計:
動物模型:
健康SD大鼠分為兩組:
對照組:僅用血管緊張素受體拮抗劑坎地沙坦(阻斷T3誘導的腎素釋放)。
T3組:T3(10μg/kg/天)+坎地沙坦,干預7周。
目的:排除RAAS激活對血壓/氧化應激的干擾。
體內檢測:
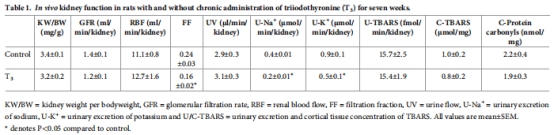
腎功能:腎小球濾過率(GFR)、腎血流量(RBF)(表1)。
氧代謝:Clark氧電極(Unisense)測量皮質/髓質氧分壓(圖1C-D)。
腎病標志物:蛋白尿、白蛋白尿(圖2A-B)、組織纖維化(圖2C)。
線粒體功能:
離體腎皮質線粒體呼吸檢測(Oroboros O2k系統)(圖3)。
排除混雜因素:
監測血糖、血壓、氧化應激(TBARS、蛋白羰基化)(表1-2)。
四、測量的數據及其研究意義
1. 腎氧代謝與缺氧(體內)
數據來源:圖1(氧耗與氧分壓)、表1(腎功能)。
關鍵結果:
T3組總腎氧耗(QO?)↑35%(圖1A),皮質氧分壓↓40%(圖1C),髓質氧分壓↑(圖1D)。
GFR、RBF無變化(表1),證實缺氧源于代謝增加而非灌注不足。
研究意義:首次在無代謝疾病模型中證明氧耗增加直接導致皮質缺氧。
2. 腎病表型
數據來源:圖2(蛋白尿/纖維化)。
關鍵結果:
T3組白蛋白尿↑3倍(圖2B),5/8動物出現腎小管間質纖維化(圖2C)。
研究意義:缺氧直接誘發腎損傷,支持“缺氧是腎病統一通路”假說。
3. 線粒體機制(體外)
數據來源:圖3(線粒體呼吸)、表2(全身參數)。
關鍵結果:
T3組線粒體解偶聯↑:UCP-2和ANT介導的質子漏增加(圖3A-B),基礎質子漏↑80%。
呼吸控制比(RCR)↑,表明ATP合成效率↑(正文)。
研究意義:T3通過增加線粒體質子漏提升氧耗,與ATP生成無關。
4. 混雜因素排除
數據來源:表2(全身參數)、表1(氧化應激)。
關鍵結果:
血糖、血壓、心臟重量無變化(表2)。
腎組織TBARS(氧化應激標志物)無差異(表1)。
研究意義:證實腎病由缺氧本身驅動,獨立于經典風險因素。
五、結論
核心結論:
T3通過線粒體解偶聯(UCP-2/ANT)增加腎氧耗,導致皮質缺氧,直接誘發腎病。
該過程獨立于高血壓、高血糖及氧化應激,為“缺氧致腎病”假說提供最直接證據。
臨床意義:
解釋甲狀腺功能亢進患者腎損傷機制(約30%出現蛋白尿)。
提示靶向線粒體解偶聯或改善腎氧供為腎病防治新策略。
六、丹麥Unisense電極數據的詳細解讀
1. 測量方法與數據位置
技術原理:使用Unisense Clark型氧電極(方法部分),實時監測腎組織氧分壓(PO?),空間分辨率達細胞水平。
檢測場景:
皮質/髓質PO?:電極直接插入腎皮質(圖1C)和髓質(圖1D),連續記錄穩態氧分壓。
校準標準:電極預校準于0%(無氧)和100%(空氣飽和)氧分壓環境。
2. 關鍵結果與機制關聯
結果:
皮質PO?:對照組≈40 mmHg → T3組≈25 mmHg(圖1C)。
髓質PO?:對照組≈15 mmHg → T3組≈20 mmHg(圖1D)。
機制關聯:
皮質缺氧:直接由氧耗增加(QO?↑)引起,與血流無關(RBF不變)。
髓質氧合改善:可能因近端小管鈉重吸收↑,減少髓袢升支粗段耗氧(討論)。
3. 研究意義
技術優勢:
高時空分辨率:實時原位監測不同腎區氧動態,避免離體檢測失真。
微創定量:優于組織染色等半定量方法,提供PO?絕對值。
科學價值:
首次在非糖尿病/高血壓模型中量化皮質缺氧程度(PO?↓15 mmHg),確立缺氧與腎病的因果關系。
揭示腎區特異性缺氧模式:皮質敏感而髓質耐受,指導靶向干預。
領域貢獻:為“腎臟缺氧理論”提供關鍵實驗工具,推動缺氧靶向治療研究。
總結:本研究通過Unisense電極等多項技術,證明甲狀腺激素通過線粒體解偶聯增加腎氧耗,導致皮質缺氧并誘發腎病,為腎病防治提供了新靶點。